
La Fenilcetonuria es una enfermedad metabólica, de tipo autosómico recesivo asociada con retardo mental. Se presenta el caso de un lactante de 9 meses de edad, diagnosticado en el período neonatal y atendido en el Centro de Atención Nutricional Infantil Antímano (CANIA), durante el año de 1999. El tratamiento nutricional consistió en dieta restringida en fenilalanina con aporte de tirosina utilizando una fórmula especial libre en fenilalanina y alimentos complementarios con bajo contenido en fenilalanina. Durante los nueve meses de seguimiento se realizaron 24 controles con un intervalo promedio de 7 a 15 días, donde intervino un equipo multidisciplinario . La intervención nutricional estuvo dirigida a mantener los niveles séricos de fenilalanina entre 2 y 6 mg/dL, lo cual requirió de ajustes continuos en las cantidades de leche materna, fórmula libre en fenilalanina y alimentos complementarios con bajo contenido de fenilalanina, iniciados a partir de los 5 meses de edad, con un aporte promedio de 30 mg/Kg/día de fenilalanina. Estos ajustes se hicieron de acuerdo a las condiciones clínicas, necesidades nutricionales, ingesta calórica y a los niveles séricos de fenilalanina. Uno de los pilares fundamentales en el éxito del tratamiento es el aspecto educativo que permite crear conciencia de la enfermedad y el entrenamiento de la madre. An Venez Nutr 2000; 13(1 ):202-209
Palabras clave: Fenilcetonuria, pediatría, nutrición.
Phenylketonuria is an autosomal recessive aminoacidopathy that leads to mental retardation when left untreated. A 9- months old infant case, who was diagnosed during the neonatal period, and treated at the Centro de Atención Nutricional Infantil Antímano (CANIA), is presented. During the 9-month period, he was seen by a multidisciplinary team. 24 follow-up visits were performed, mean period time between visits were 7 to 15 days.
Treatment was aimed to maintain the plasma phenylanine between 2 -6 mg/dL and consisted of a diet low in phenylanine by supplementation with phenylanine- free formula and complementary food low in phenylanine started at 5 months with a mean phenylalanine intake of 30 mg/kg/day.
Thequantity of breast milk, phenylanine- free formula and complementary food was altered according to the plasma phenylanine, clinical conditions, calorie intake and nutritional needs.
An improved knowledge and understanding about this disease may lead to improved dietary compliance and parental training and counseling is a pivotal feature in treatment success. An Venez Nutr 2000; 13(1 ):202-209
Key words: Penylketonuria. Pediatrics. Nutrition.
La Fenilcetonuria es una enfermedad metabólica de tipo autosómico recesivo asociada con retardo mental, en la cual la conversión de fenilalanina en tirosina está alterada por déficit o ausencia de la enzima hepática Fenilalanina Hidroxilasa (FH).
La enzima Fenilalanina Hidroxilasa está codificada en el cromosoma 12q22-q24.1, las hiperfenilalaninemias se producen por una gran variedad de mutaciones a este nivel, en la actualidad se han descrito más de trescientas mutaciones a escala mundial; en 1998 se publicó un artículo sobre las bases moleculares de la fenilcetonuria en Venezuela donde se describen dos nuevas mutaciones (1).
El exceso de fenilalanina en el organismo es tóxico, e interfiere con el normal desarrollo y maduración del sistema nervioso, produciendo lesiones irreversibles que conducen a retardo mental. Los pacientes no tratados o diagnosticados tardíamente tendrán una reducción del coeficiente intelectual en un 95% de los casos. Se estima que una persona entre 50 es portadora de un gen mutante y la prevalencia mundial de la enfermedad es 1 en 12000 a 15000 recién nacidos. La prevalencia varía con la región geográfica y con el grupo étnico 12).
En nuestro país se estima que las cifras son mucho más bajas y similares a las del grupo de México, aproximadamente 1 en 70000 recién nacidos vivos(3).
En Venezuela la mayoría de los niños con fenilcetonuria han sido diagnosticados tardíamente. Entre los años 1965 y 1970 en el Hospital "J.M. de los Ríos" se evaluaron 4675 pacientes menores de 5 años encontrando 9 casos positivos para fenilcetonuria, todos diagnosticados después de los 6 meses de edad(4).
La prueba de descarte neonatal para fenilcetonuria se realiza en Venezuela desde 1985(3). Entre Octubre de 1985 y julio de 1992 se realizó la prueba a 15.726 recién nacidos, identificándose 3 casos sospechosos de hiperfenilalaninemia; uno de ellos falleció antes de repetir la prueba y los otros dos resultaron negativos(5). Entre 1994 y 1998 se realizaron 86.599 pruebas, de las cuales resultaron sospechosas 190 y solo un caso confirmado en 1998(3), que en el caso que presentamos.
En la consulta de Génetica Humana del Instituto Venezolano de Investigaciones Científicas (IVIC), se han estudiado alrededor de ocho familias de casos índice de Fenilcetonuria, de las cuales solo tres se consideran casos autóctonos, determinándose un foco epidemiológico para la enfermedad ubicado en el Municipio Pedro Zaraza entre el estado Guárico y Anzoátegui(6).
Criterios Diagnósticos de fenilcetonuria clásica(7):
Nivel de Fenilalanina en sangre> de 16 a 20 mg/ 100ml
Nivel de Tirosina en sangre menor de 3 mg/ 100ml
Presencia de Acido Fenilpirúvico y/o- hidroxifenilacético en orina
El tratamiento nutricional adecuado consiste en una dieta restringida en fenilalanina con un aporte de tirosina que permita mantener los niveles séricos de fenilalanina entre 2 y 6 mg/dL(7-9) y un crecimiento y desarrollo normal.
Por lo anteriormente expuesto la información nacional que se tiene sobre el manejo temprano de estos niños es escasa, por lo que consideramos de gran valor, compartir la experiencia del manejo de este paciente.
1. Caso clínico
Lactante masculino de 28 días de vida referido de la Unidad de Estudios de Errores Innatos del Metabolismo (UNIDEIM) del Instituto de Estudios Avanzados (IDEA) por presentar en la prueba de descarte neonatal, valores elevados de fenilalanina y un valor sérico mayor a 20 mg/dL de fenilalanina en la prueba confirmatoria con niveles normales de los aminoácidos restantes. Diagnóstico de referencia: Hiperfenilalaninemia en estudio.
1.1. Antecedentes encontrados en la primera evaluación:
Perinatales: Producto de primera gesta, embarazo controlado desde el tercer mes, sin complicaciones, a término (38 semanas), parto espontáneo, Peso: 2700 g. Talla: 47 cm. Presentó hipoxia perinatal e ictericia por lo que permaneció 12 días en Unidad de Cuidados Neonatales. Diagnóstico neonatal: RNAT/AEG.
Alimentación: lactancia materna exclusiva
Familiares: Madre de 23 años, aparentemente sana, procedente de Caracas. Procedencia de la familia materna: Abuela de Ocumare del Tuy, Abuelo de Barcelona. Prima materna con retardo mental.
Padre aparentemente sano procedente de Caracas. Familia paterna procedente de Caracas
Consanguinidad: negativa
Sociales: Familia en pobreza extrema según método de Graffar Modificado.
Nivel Educativo de la madre: educación básica completa.
1.2. Examen físico:
Buenas condiciones generales, fenotípicamente normal, en el examen físico solo resaltan lesiones tipo pápulas y eritema en área del pañal.
1.3. Antropometría: (Cuadro 1)
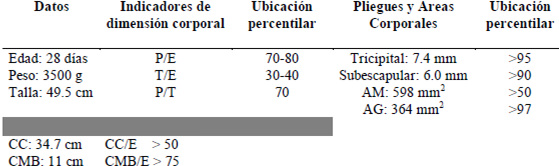
1 .4. Paraclínico:
Exámenes de laboratorio: hematología y química sanguínea dentro de los parámetros normales.
Test de hipotiroidismo congénito: negativo
1.5. Diagnóstico Nutricional lntegral y asociado de ingreso:
lactante eutrófico.
Fenilalaninemia en estudio.
Dermatitis área del pañal.
Hábitos de alimentación adecuados. Apetito Bueno.
Conducta de alimentación adecuada.
1.6. Plan Inicial:
Se indicó fórmula libre en fenilalanina la cual no se comercializa en el país, por lo que se debió gestionar su adquisición a través de la intervención del Trabajador Social. Hasta el momento en que se dispuso de la fórmula, el paciente se mantuvo con lactancia materna exclusiva. Se le brindó información a la madre sobre la patología y manejo dietético de la misma.
Se inició intervención del Psicólogo para apoyo, orientación por tratarse de una patología crónica.
Estudio metabólico confirmatorio en orina de 24 horas. Consejo genético.
2.7. Método de trabajo:
Durante los nueve meses del seguimiento se realizaron 24 controles, con un intervalo promedio de 7 a 15 días. En cada uno de estos controles el paciente fue evaluado por el
Pediatra - Nutrólogo y la Nutricionista Clínico a través de la siguiente rutina:
Para los indicadores de dimensión global se utilizaron los valores de referencia de la OMS (puntos de corte percentil 10 y 90) Y para los indicadores de composición corporal se utilizó los valores de referencia del Estudio Transversal Caracas (puntos de corte percentil 10 y 90).
Cálculo del requerimiento de energía y nutrientes El cálculo del requerimiento de Energía se realizó a través del método fraccionado(11). Con este método el requerimiento energético estimado se mantuvo alrededor de 127 Kcal/kg/día y en los períodos de intercurrencia considerando el gasto energético por catabolismo (10 a 20 %), se elevó a 146 Kcal/kg/día.
Para el cálculo del requerimiento de proteínas y de fenilalanina se revisaron las diferentes recomendaciones sugeridas en la literatura de referencia (Cuadro 2 y 3); el aporte promedio de proteínas utilizado en el paciente durante los primeros 6 meses, fue de 4.4 g/kg/día y 3.2 g/kg/día a partir del sexto mes.


Para calcular el aporte diario aproximado de fenilalanina, se obtuvo el estimado de la cantidad de leche materna que el paciente recibía pesando al niño antes y después de haber recibido leche materna y tomando en cuenta la duración y frecuencia de las tomas. Conociendo que 1 gramo de leche materna aporta 0.43 mg de fenilalanina (12,14) se calculó el aporte total.
Con respecto al aporte de fenilalanina, hasta antes de iniciar la incorporación de alimentos complementarios, se vigiló que el aporte de fenilalanina se mantuviera dentro del rango recomendado para la edad; posteriormente, se trabajó con un aporte promedio de 30 mg/kg/día. Con el objetivo de mantener los niveles de fenilalanina en sangre entre 2 y 6 mg/dL(7,9), se indicó la cantidad de fórmula o de leche materna dependiendo de los resultados de los niveles séricos de fenilalanina encontrados para cada evaluación.
Una vez que se inició la incorporación de alimento complementarios, se trabajó con la madre lo referente a: Pesada y medida de los alimentos. Nociones de grupos de alimentos, listas de sustitutos por grupos de alimentos de bajo contenido de fenilalanina las cuales fueron elaboradas tomando en cuenta los alimentos de consumo habitual local.
Utilización del plan de alimentación y distribución de alimentos a lo largo del día.
Preparaciones de alimentos utilizando la fórmula libre de fenilalanina para incorporar nuevos sabores y texturas.
Diario de ingesta con un formato previamente elaborado en donde se registra la cantidad en gramos de alimentos consumidos y dejados en el plato.
2.8. Evolución:
Una vez que se dispuso de la fórmula libre en fenilalanina y teniendo en cuenta que los niveles de fenilalanina en sangre estaban elevados (> de 20 mg/dL), se suspendió temporalmente (10 días) la lactancia materna, y se aportó el 100 % del requerimiento calórico total con la fórmula especial libre en fenilalanina. Posteriormente, se asumió que los niveles de fenilalanina se encontraban en el rango deseable y se reinició la lactancia materna y la fórmula libre en fenilalanina se mantuvo aportando el 80% del requerimiento de proteínas(9).
La Intervención dietética en las posteriores consultas dependió de las condiciones clínicas, las necesidades nutricionales, la ingesta dietética y los niveles séricos de fenilalanina. En varias oportunidades no contamos con los resultados de fenilalanina en el momento de la consulta ya que la muestra para este análisis era tomada en nuestro Centro y posteriormente enviada a IDEA Instituto de nuestra ciudad donde se procesa ésta.
Dependiendo de las variables anteriormente señaladas, se realizaron los ajustes correspondientes para la lactancia materna, fórmula especial libre en fenilalanina y alimentos complementarios como se resume en el (Cuadro 4).
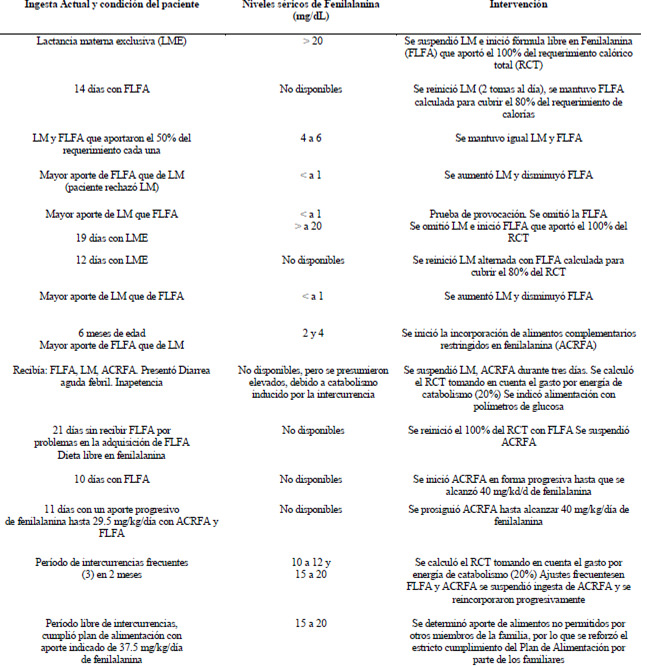
La evolución desde el punto de vista clínico se puede dividir en tres períodos. Durante el primer período (1 a 3 meses de edad), no se registró ninguna intercurrencia, el desarrollo psicomotor era adecuado, aunque se evidenció hipertonía muscular. Las velocidades de peso y talla se ubicaron por encima del percentil cincuenta y fue el lapso donde se logró el mejor control de los niveles séricos de fenilalanina (> de 20 mg/dL al inicio y luego entre 2 y 6 mg/dL).(Cuadro 2).
El rápido descenso de los niveles de fenilalanina y su mantenimiento inclusive con un aporte predominante de lactancia materna, llamaba la atención y se decidió realizar una prueba de provocación que nos permitiría hacer diagnóstico diferencial con hiperfenilalaninemias transitorias, posterior a 11 días de lactancia materna exclusiva los niveles se elevaron sobre 20 mg/dL, lo que apoyaba el diagnóstico de fenilcetonuria clásica.
En el segundo período (3 a 6 meses de edad), se presentaron tres intercurrencias infecciosas, dos respiratorias y una gastrointestinal. Su desarrollo psicomotor progresaba en forma adecuada, la hipertonía muscular se generalizó predominando en extremidades, con mayor compromiso del tono activo y movimientos poco controlados, por lo que se refirió al Centro de Desarrollo Infantil. Durante este período, se continuó el plan de estimulación para el hogar en las distintas áreas del desarrollo indicado por Psicología desde los 2 meses de edad.
Los niveles séricos de fenilalanina se ubicaron fuera de los valores deseados en tres oportunidades, incluyendo cifras de 15 a 20 mg/dL en dos de éstas, durante las cuales se observaron lesiones en piel tipo eczematosas en mejillas, tronco y área del pañal. Se reciben los resultados de presencia de Ácido Fenilpirúvico y hidroxifenilacético en orina, confirmando el diagnóstico de fenilcetonuria clásica.
Las velocidades de peso y talla se ubicaron en el percentil 75 y entre los percentiles 90 y 97 respectivamente. En el último período (6 a 9 meses de edad), se presentaron tres intercurrencias infecciosas, dos respiratorias, una gastrointestinal y una convulsión febril. En este período es evaluado en su tercer control por Neuropediatría su desarrollo psicomotor se mantenía acorde a su edad cronológica, se indico Diazepan SOS en caso de fiebre, solicitó Resonancia Magnética Nuclear cerebral y Electroence- falograma.
Los niveles de fenilalanina se mantuvieron elevados la mayor parte del tiempo, ( 10 a 15 mg/dL hasta 15 a 20 mg/dL), incluso una vez superadas las intercurrencias, lo cual se relacionó con transgresiones dietéticas por parte del grupo familiar, por lo que se hizo necesario reforzar la intervención de Psicología, con la finalidad de trabajar conciencia de enfermedad en el grupo familiar. Para el fínal de este período las velocidades instantáneas de peso y talla entre el sexto y noveno mes descendieron por debajo del percentil tres y diez respectivamente. Sin embargo se mantiene eutrófico pero su talla se ubicó en zona crítica negativa.
No fue necesaria la suplementación con micronutrientes, ya que los mismos fueron aportados por la fórmula libre en fenilalanina en forma adecuada; se vigiló especialmente las adecuaciones de calcio, hierro y zinc(14).
El principal factor de éxito en el tratamiento de esta enfermedad es el diagnóstico precoz a través de la prueba de despistaje neonatal, la cual en Venezuela no es de práctica obligatoria. En la interpretación de los resultados de esta prueba es importante considerar el momento en cual se realiza la misma, las condiciones clínicas y el tipo de alimentación del paciente, ya que pruebas realizadas en las primeras 24 horas de vida pueden resultar en falsos negativos y es recomendable entonces repetirla a partir de las 72 horas de vida(15,16). En concordancia con estas observaciones, en nuestro país, a partir de 1997 se decidió aplicar la prueba de descarte neonatal a partir del cuarto día de lactancia materna, realizándose entre 1997 y 1998 aproximadamente 20.000 pruebas con un solo caso confirmado, es posible que con esta nueva normativa, aumente el número de casos diagnosticados (2).
Los pacientes con fenilcetonuria no tienen características fenotípicas específicas pero hay síntomas y signos que pueden observar, tales como: eczemas, microcefalia, temblores, movimientos espásticos en extremidades, posturas inusuales, convulsiones, hiperactividad, retardo en adquisición de destrezas mentales y sociales, olor característica ratón de la orina y sudor y coloración clara del cabello ojos(13,17,18). Las lesiones en piel observadas en la primera evaluación e interpretadas como una dermatitis del área del pañal, reaparecieron en las diferentes oportunidades en que los niveles de fenilalanina se elevaron. Lo cual coincide con las lesiones de tipo eczematosas reportadas en la literatura(13,18). De igual modo la convulsión disparada por fiebre que presentó el paciente ha sido descrita como uno de los signos de la enfermedad(18).
La suspensión total del aporte de fenilalanina en la dieta, se realizó de acuerdo a lo recomendado por la literatura cuando los niveles de fenilalanina en sangre se encontraron por arriba de 15 mg/dL(8,13).
La velocidad promedio de descenso de fenilalanina en sangre resultó entre 1.8- 2 mg/dL/día, muy por debajo de lo reportado en la literatura 5 -10 mg/dL/día y 5 a 9 mg/dL/día(8). Este valor fue tomado en cuenta posteriormente para el estimado del número de días necesarios para lograr valores deseables.
La prolongada suspensión del aporte de fenilalanina reduce a cifras menores a 2 mg/dL, con riesgo de comprometer el estado nutricional (desnutrición protéico calórica)(18), mientras que, los niveles elevados serían responsables del deterioro neurológico, no solo por el efecto tóxico de la fenilalanina en el sistema nervioso, sino también por la inhibición competitiva que producen estos altos niveles de fenilalanina respecto a otros aminoácidos (metionina, triptófano, histidina, tirosina, isoleucina, leucina y valina)(17). El cumplimiento de esto en la práctica resulta difícil y en este caso no siempre fue posible, a pesar de que se realizó un monitoreo sistemático del paciente (controles semanales o llamadas telefónicas para notificar intercurrencias) durante la intervención; las consecuencias de estas elevaciones aunque transitorias fueron fuente de preocupación ya que estudios en vivo tanto en ratas como en humanos han demostrado que no solo elevaciones a largo plazo sino también picos de concentraciones plasmáticas de fenilalanina pueden alterar la función mental(19). En el gráfico 1 se muestran las variaciones de los niveles séricos de fenilalanina y los episodios de intercurrencias, mostrando clara relación entre estos últimos y niveles elevados de fenilalanina.
Se ha descrito un ritmo circadiano en los niveles de fenilalanina en sangre con valores más altos durante la mañana y más bajos en la tarde y noche, también se ha encontrado una gran variabilidad en los niveles séricos de fenilalanina a lo largo del día en niños con fenilcetonuria que presentan niveles matutinos aceptable de fenilalanina, la significancia de este hecho no se conoce y se piensa que está en relación con el consumo diario total del sustituto protéico a lo largo de un período de 10 horas(20). Por lo tanto, se vigiló estrictamente que la fórmula libre de fenilalanina y los alimentos que aportan fenilalanina se distribuyeran en igual proporción a lo largo del día(8,19); el entrenamiento a la madre fue determinante para el cumplimiento de este aspecto.
La pérdida del apetito durante el período de las intercurrencias, como era de esperar, se presentó conjuntamente con la disminución de la ingesta calórica aportada tanto por la fórmula especial libre en fenilalanina como de los alimentos con aporte en fenilalanina por lo que fue necesario en varias oportunidades suplementar la alimentación con módulos de polímeros de glucosa. El período de intercurrencia trae consigo un aumento en el catabolismo protéico ya sea por el consumo insuficiente de energía o por la intercurrencia misma, lo cual conduce a elevaciones de los niveles de fenilalanina. Contribuye a esta elevación el uso de medicamentos que contienen fenilalanina, por lo tanto es necesario conocer la composición de estos productos, concentración de fenilalanina y tomarlo en cuenta en el cálculo del aporte diario total(21).
La orientación anticipada sobre el manejo de estas situaciones es altamente recomendable a fin de prevenir el catabolismo tisular y mantener la ingestión de la fórmula tanto como sea posible (7).
El inicio precoz del tratamiento y control adecuado del niño con fenilcetonuria garantiza un crecimiento adecuado respecto a su potencial genético y le permite alcanzar una talla normal(22). Los protocolos iniciales de tratamiento que recomendaban mantener niveles plasmáticos de fenilalanina entre 1 a 3 mg/dL se asociaron a alteraciones del crecimiento y retardo en la maduración esquelética en muchos pacientes(23). Con la recomendación posterior de mantener niveles plasmáticos entre 2 a 6 mg/dL se han garantizado velocidades de crecimiento y maduración esquelética normales. La afectación de las velocidades de peso y talla en este paciente se presentaron en el período donde los niveles de fenilalanina séricos se mantuvieron elevados en la mayor parte del tiempo.
La importancia del compromiso con el tratamiento por parte del cuidador está en que éste es para toda la vida, estudios prospectivos en pacientes que abandonaron la dieta a los 6 años, 8 años y 12 años presentaron descensos en el coeficiente intelectual. En niños mayores, la hiperfenilalaninemia produce cambios conductuales y déficit de atención, sintomatología que se revierte cuando se instaura nuevamente la dieta especial (14). Así mismo la vigilancia del nivel de desarrollo y nivel intelectual se debe mantener durante todo el tratamiento con la aplicación de escalas de desarrollo infantil y mediciones de nivel intelectual.
El manejo del paciente con fenilcetonuria debe ser realizado por un equipo multidisciplinario que interactúe en forma directa y continua con el cuidador y el grupo familiar del paciente a fin de garantizar una mejor calidad de vida y evitar el retardo mental.
Constituyen elementos claves del tratamiento, la información sobre la enfermedad y sus consecuencias de una manera sencilla y comprensible así como la educación nutricional, especialmente en referencia al tratamiento dietético, en donde la madre se entrena para conocer los grupos de alimentos, aportes de fenilalanina de los mismos, método de recolección de ingesta y manejo en situaciones de enfermedad. La fenilcetonuria es una enfermedad de baja prevalencia en Venezuela, por lo que la experiencia en el tratamiento de la misma es escasa y las principales limitaciones que se plantean, son la dificultad para obtener la fórmula especial libre en fenilalanina en el mercado nacional, su alto costo de importación, y mantener un seguimiento estricto del paciente y su grupo familiar.
El aspecto educativo entendido como la información sobre la patología que permita crear conciencia de la enfermedad y el entrenamiento que debe recibir la madre o cuidador de estos pacientes, constituye uno de los pilares fundamentales en tratamiento.
En el área de la investigación, se hace necesario trabajar en la incorporación del contenido de fenilalanina y otros amino- ácidos en las tablas de composición de alimentos de nuestro país, a fin de permitir dietas mas adaptadas, no tan solo para el tratamiento de la fenilcetonuria sino también para otras enfermedades metabólicas. De igual manera es necesario plantear, la creación de un mecanismo que facilite la adquisición de las fórmulas especiales, productos insustituibles para el tratamiento en este tipo de enfermedades.
El establecimiento de grupos de apoyo para las familias de pacientes con enfermedades metabólicas ha demostrado ser una herramienta útil, por lo que su promoción sería recomendable.
Finalmente se debe establecer y promocionar centros de referencia y asociaciones estratégicas entre los distintos Centros que reciben pacientes con enfermedades metabólicas, así como el intercambio de información que permita desarrollar protocolos locales de tratamiento.