
Desde 1988 se describe el síndrome metabólico (SM) como una forma de identificar a las personas con alto riesgo de enfermedad cardiovascular y se propone que la obesidad es indispensable para el diagnóstico del SM La industrialización permitió cambios en el estilo de vida: una disminución del ejercicio físico y el consumo de una dieta alta en calorías, rica en grasa y pobre en fibra dietética. Desde el punto de vista evolutivo el hombre desarrolló un genotipo ahorrador, que asegura los cambios metabólicos necesarios para acumular glucógeno muscular y triacilglicéridos en el tejido adiposo durante los períodos de escasez. Cuando esta carga genética se somete a períodos de abundancia puede provocarse un conjunto de trastornos que favorecen la aparición de enfermedades crónicas no trasmisibles. Así se establecen los criterios para el diagnóstico de SM: Obesidad abdominal, hipertrigliceridemia, disminución plasmática de HDL y aumento de glucemia. En esta revisión se destaca la importancia de la obesidad en la aparición de enfermedad cardiovascular (ECV) y el síndrome de resistencia a la insulina para abordar el SM. Respecto al diagnóstico de SM se discuten los cambios en los puntos de corte para las variables diagnósticas y la posible importancia de esta modificación. Se incluyen comentarios sobre los mecanismos fisiopatológicos propuestos para el desarrollo de este síndrome, y se hace énfasis en la importancia del diagnóstico y estudio del SM como elemento preventivo de ECV. An Venez Nutr 2007;20 (2): 92-98.
Palabras clave: Síndrome metabólico, obesidad, genética, resistencia a la insulina, enfermedad cardiovascular, circunferencia abdominal.
Metabolic Syndrome (MS) was described in 1988 to identify high cardiovascular risks patients, being obesity an important component of this syndrome. Industrialization allowed changes in life style: lower physical activity, a change in the dietary pattern leading to a high calorie, high fat diet with a low dietary fiber content. From an evolutionary point of view “thrifty” genes were selected at a time when food resources were scarce, to grant the necessary metabolic changes in order to accumulate muscular glycogen and triglycerides in adipose tissue during difficult periods. It was proposed that these adaptations only became detrimental when this genetic pool is confronted with a nutrient abundant environment, which leads to an increased risk to develop non transmissible chronic diseases. The diagnostic criteria for Metabolic Syndrome are: Abdominal obesity, hipertriglyceridemia, low plasma HDL and hyperglycemia. This review stresses the importance of abdominal obesity in the generation of cardiovascular disease and also insulin resistance in the metabolic syndrome. A reference to discrepancies in the cutoff values of different variables used to diagnose the MS is made and its importance is discussed. Brief comments on the pathophysiology of the MS are included and the importance of the diagnosis and further research on MS in order to prevent cardiovascular disease is mentioned. An Venez Nutr 2007;20 (2): 92-98.
Key words: Metabolic syndrome, obesity, genetics, insulin resistance, cardiovascular disease, abdominal circumference.
Solicitar copia a: uglive@cantv.net
El síndrome metabólico fue descrito en 1988 por Reaven (1) como una forma práctica de identificar a las personas que requieren hacer un cambio de vida para disminuir su riesgo de enfermedad cardiovascular. La presencia de obesidad se considera indispensable para diagnosticar dicho síndrome. En este sentido, la evolución del Homo-Sapiens desde la era paleolítica hasta nuestros días muestra que en el paleolítico, para conseguir alimentos, el hombre necesitaba realizar una enorme actividad física diaria, a diferencia de la época actual, cuando el progreso tecnológico y la industrialización permiten almacenar alimentos y tenerlos “a mano” para su preparación y consumo. Este cambio de estilo de vida provocó varios efectos: el consumo de una dieta baja en fibra dietética y rica en grasas y alimentos refinados; una disminución del ejercicio físico y un aumento en el peso corporal. Todos ellos moduladores muy importantes de la secreción de insulina (2).
Como consecuencia de la interacción entre los cambios de actividad física y los procesos metabólicos ocurridos en el hombre del paleolítico (50,000 a 10,000 A.C.), se favoreció la aparición de un genotipo ahorrador al seleccionarse los genes que aseguran una maquinaria enzimática capaz de generar una buena reserva de glucógeno muscular y de triacilglicéridos en el tejido adiposo (TA), que son sustratos requeridos para sobrevivir en un hábitat hostil. Es decir que los ciclos de actividad- reposo, alimentación - ayuno, incidieron en el proceso metabólico, modificándose la expresión del pool genético favoreciendo los procesos bioquímicos que permiten conservar, restituir y almacenar el glucógeno muscular. Neel (3) propuso el término de genes ahorradores para referirse a esta selección de un genotipo que asegure por una parte la eficiente utilización y almacenamiento de la energía consumida durante los períodos de abundancia y por otra la sobrevida de la especie, gracias a esa reserva energética acumulada durante los períodos de escasez.
Esta dotación genética, no modificada al menos desde hace 10.000 años, debió enfrentar hace menos de 200 años un patrón distinto de disponibilidad de alimentos. Con la industrialización aparecen alimentos en abundancia, que se caracterizan por poseer una alta densidad calórica y un bajo contenido en fibra dietética, y al mismo tiempo la actividad física deja de ser un factor esencial para conseguir estos recursos. El consumo calórico aumentado y la presencia de genes ahorradores que permiten el uso eficiente de esta energía pueden explicar el aumento de peso y la predisposición a la aparición de enfermedades crónicas no transmisibles como obesidad, diabetes tipo 2 y síndrome metabólico (4,5).
La hipótesis del “Fenotipo ahorrador” (6,7) plantea que la subnutrición tanto intrauterina como infantil, es capaz de programar una expresión génica para favorecer la sobrevida. Se protege al cerebro frente a otros tejidos, lo que conduce a una programación metabólica condicionada por la escasez del sustrato bioenergético. Los modelos experimentales de malnutrición intrauterina, (8-10), y en particular de déficit protéico, han demostrado claramente una alteración en la estructura-función del páncreas con disminución de las células â de los islotes de Langerhans (11) y una menor capacidad secretora de insulina (12). Esta programación celular afecta igualmente la regulación del gasto energético (13-15).
De acuerdo a esta hipótesis la subnutrición, que se caracteriza por un bajo peso al nacer, provocaría una programación in utero que explicaría la aparición de la mayor parte de la obesidad, el síndrome metabólico y la diabetes mellitus tipo 2 en un adulto que nació con bajo peso para la edad gestacional y que luego ha sido expuesto a un ambiente de abundancia alimentaria (15-17). Aún más, las evidencias de numerosos estudios indican que esta programación no se limita a la primera generación (8,18).
Los estudios epidemiológicos realizados en personas que se supone poseen genotipo ahorrador (Indios Pima) y en grupos humanos con Fenotipo Ahorrador, como las personas sometidas a desnutrición aguda por hambruna, (población de Holanda durante la ocupación de la Alemania Nazi), aportan evidencias que sustentan ambas hipótesis (19,17).
Es indudable que el peso corporal del adulto y en particular la masa de tejido adiposo es el resultado de la interacción entre los factores genéticos y los ambientales. Los genes asociados con la presencia de obesidad y del síndrome metabólico incluyen varios grupos:
Visto así, el hombre moderno es el producto de confrontar un “viejo genoma” con un nuevo medio ambiente que ofrece exceso de nutrientes que, al ser ingeridos, pueden almacenarse eficientemente en el tejido adiposo (TA) y como resultado aparecen la obesidad (26) y el riesgo aumentado a sufrir enfermedades degenerativas crónicas (27,28).
Como resumen muy simplista podemos decir que a priori, poseer una carga genética que condicione a la delgadez, es un factor de protección contra la obesidad y el síndrome metabólico y que en contraposición las evidencias epidemiológicas y experimentales indican que la malnutrición por déficit durante las primeras etapas de la vida, particularmente en el período fetal, aumenta el riesgo a desarrollar obesidad y síndrome metabólico cuando se asegura el libre acceso a una dieta inadecuada y rica en calorías.
La obesidad es el aumento en la acumulación de tejido adiposo (TA) que cursa con aumento del peso corporal. La plasticidad del TA es la clave para entender la aparición de obesidad, pues el adipocito se comporta como un tejido dinámico clave en la respuesta a la adaptación nutricional aumentando la producción de citoquinas (29) y sufriendo una desregulación caracterizada por un aumento de su capacidad para expandirse (30).
El indicador de obesidad mundialmente reconocido es el Índice de Masa Corporal (IMC) cuyo cálculo es sencillo, por lo cual es usado frecuentemente para clasificar las modificaciones del peso corporal de un sujeto.
El IMC se calcula dividiendo el peso del sujeto, en kilogramos, por el cuadrado de la talla expresada en metros (IMC = Peso (kg) / Talla (m2); sin embargo, se debe tomar en cuenta que el IMC puede aumentar en individuos que tienen una gran masa muscular (atletas, fisioculturistas), o una masa ósea aumentada (acromegalia). En estos casos el IMC aumentado no se corresponde con el sobrepeso o la obesidad.
Desde hace mucho tiempo se ha descrito que la obesidad aumenta el riesgo de aparición de ciertas enfermedades y que el incremento del diámetro de la circunferencia abdominal podría ser usado como indicador de obesidad.
El Cuadro 1 muestra los riesgos relativos de enfermedad en función del IMC y de la circunferencia abdominal (CA).

Del Cuadro anterior se puede deducir que:
Uno de los efectos secundarios del sobrepeso y la obesidad es la aparición de un aumento en los niveles plasmáticos de insulina, que en los estadios iniciales cursa con valores de glucemia dentro de límites normales (31).
El síndrome de resistencia a la insulina no es una enfermedad sino un término usado para describir un proceso fisiopatológico que se caracteriza por una disminución de la sensibilidad tisular a la acción de la hormona, lo cual provoca la respuesta homeostática compensadora aumentando la producción de insulina (32). A la larga esta elevación de los niveles plasmáticos de hormona acompañada de la disminución de la utilización periférica de la glucosa puede generar un aumento de la glucemia e inducir una disfunción metabólica que puede provocar serias consecuencias clínicas entre las cuales se incluyen Diabetes Mellitus tipo 2 (DM2) (33), enfermedad cardiovascular (ECV), algunas dislipidemias -en particular hipertrigliceridemia-, síndrome de ovario poliquístico e hipertensión arterial entre otras (34).
Este síndrome se caracteriza por la presencia simultánea de un conjunto de factores de riesgo – obesidad, hipertrigliceridemia, hiperglucemia e hipercolesterolemia entre otros - para desarrollar Enfermedad Cardiovascular (ECV). Cuando son parte del síndrome metabólico, estas patologías comparten algunos rasgos etiopatogénicos comunes como son la obesidad visceral y la aparición de resistencia a la insulina. Un aspecto fundamental de la obesidad que se asocia con el riesgo a la aparición del síndrome metabólico es la distribución preferencial del TA. En general se describen dos grandes tipos de distribución del tejido adiposo:
Esta diferencia se relaciona con las características metabólicas del TA de la región intra-abdominal (33,36,35), debido a que este tejido adiposo posee una elevada actividad lipolítica, que aumenta el flujo de ácidos grasos libres en plasma (37,26). Por tanto, se aumentan los sustratos para la síntesis hepática de lipoproteínas ricas en triglicéridos (VLDL). Al mismo tiempo, no se inhibe eficazmente la producción hepática de glucosa, el músculo disminuye su captación lo cual conduce a un incremento en la glucemia, que provoca un aumento en la secreción de insulina y eventualmente hiperinsulinismo (38,39). Otra alteración que forma parte del Síndrome Metabólico es la aparición de lipoproteínas de baja densidad (LDL) pequeñas y densas (40) que siguen estudiándose como factor etiopatogénico de la aterogénesis.
La importancia de hacer el diagnóstico de Síndrome Metabólico radica en que cuando está presente en un paciente es indicador de un elevado riesgo cardiovascular. Por otra parte, cuando coexisten solamente algunos de los factores simultáneamente (no se cumplen todos los criterios para diagnosticar el síndrome) es un alerta para mantener la vigilancia del paciente.
Los Cuadros 2 y 3 presentan los diferentes criterios que se han usado para diagnosticar Síndrome Metabólico.


Posteriormente la IDF propone una modificación al disminuir los puntos de corte para la circunferencia abdominal con el propósito de asegurar que el diagnóstico de SM se base en la presencia de TA abdominal (Cuadro 3) y además señala que debe tomarse en cuenta el origen étnico de la persona (43).
La importancia de este cambio en los valores de la circunferencia abdominal aún está en discusión pues se aumenta la prevalencia del SM y no está claro que aumente el valor pronóstico de enfermedad cardiovascular en todas las poblaciones (43, 45,37).
La revisión de la literatura muestra que, a diferencia de lo que ocurre en los países desarrollados, no hay consenso en cuanto a los valores de referencia para la circunferencia abdominal de las poblaciones indoamericanas, como la nuestra. En el Cuadro 4 se indican los valores para los distintos países.
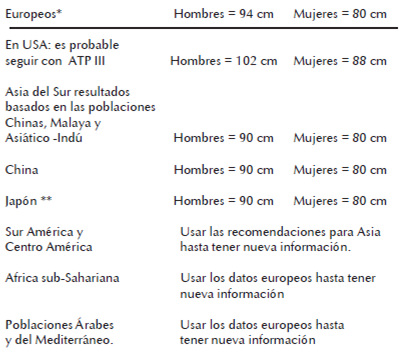
Es interesante señalar que:
Además Reaven (52) señala con insistencia que es necesario recordar que:
La fisiopatología del Síndrome Metabólico aún está en estudio. Se propone que la mayor actividad lipolítica de la grasa abdominal provoca aumento de la síntesis de triacilglicéridos. La acumulación de ácidos grasos de cadena larga en el hígado, músculo y algunas áreas cerebrales sumada a la toxicidad pancreática ejercida por estos ácidos grasos constituye factores fundamentales en las modificaciones metabólicas que permiten la aparición de este síndrome (46).
Como parte del modelo se sugiere que el aumento de los ácidos grasos puede provocar: disminución de la captación periférica de glucosa al inhibir la unión de los transportadores Glut-4 a la membrana celular, disminución de la secreción de insulina por las células β del páncreas (efecto lipotóxico) y aumento de la síntesis de triglicéridos hepáticos y VLDL que condicionan la generación de LDL pequeñas y densas (40).
Aún hay un largo camino por recorrer. Para dar un ejemplo se puede citar el trabajo de Raikkonen et al, quienes recientemente publicaron un estudio de seguimiento durante 15 años que sugiere una correlación positiva entre la presencia de factores socio ambientales como el estrés crónico y la depresión con la aparición posterior del Síndrome Metabólico (47).
Hay otros aspectos importantes de este síndrome como son la enfermedad inflamatoria y la posible participación del sistema inmunológico en esta patología (48- 50) pero su discusión no es objeto de este trabajo.
Además de ser una herramienta para el personal de salud, el concepto de Síndrome Metabólico, permite:
Resulta evidente la necesidad de continuar investigando a fondo en esta área con el propósito de llegar por una parte a un consenso para el diagnóstico y por otra a conocer los mecanismos fisiopatológicos del Síndrome Metabólico, así se podrán implementar los programas educativos que eviten su aparición y por tanto disminuirá el riesgo de aparición de enfermedades cardiovasculares. En este sentido Slentz et al (53) han demostrado que como medida preventiva además de una dieta balanceada que cubra los requerimientos calóricos de la persona, un programa de ejercicios moderado evita la acumulación de grasa abdominal y por tanto también disminuye la probabilidad de aparición de esta patología.
Recibido: 25-04-2007
Aceptado: 25-08-2007